
The new Medical Devices Regulation 2017/745 (MDR) was supposed to take full effect in Europe by mid-2020, being a fundamental revision of existing Medical Device Directive 93/42 EEC (MDD) and Active Implantable Medical Devices Directive 90/385/EEC (AIMD); following the SARS-CoV-2 outbreak in Europe, on April 23rd 2020, the EU legislator published an amendment to the EU MDR (Amending Regulation), postponing the application of most of its provisions by one year, until May 26th, 2021.
The aim of the MDR is to establish a robust, transparent, predictable and sustainable regulatory framework for Medical Devices (MDs) including combinations of MDs with In Vitro Diagnostic (IVDs) Medical Devices and/or Medicinal Products across EU member states.
All new MDs deemed to be placed in the EU member states market will have to conform to the requirements of the MDR by the end of May 2021 on.
There are 15 key major changes in the MDR versus the previous regulatory framework defined by the MDD:
- The MDR, as a regulation, carries mandatory jurisdiction that is directly applicable and enforceable in all EU Member States, while the directives are just legislative frames that set out the rules which must then be transposed into national legislation in order to become effective.
- The new regulation is four times outstretched in number of pages and number of articles and has five more annexes than its predecessor, the MDD.
- There is a big emphasis on patient’s health and safety outlined in the MDR, the word "safety" appears 290 times in the MDR, while the MDD uses it only 40 times, moreover the MDR introduces provisions for transparency and MD traceability in order to improve health and safety.
- MDR requires MDs manufacturers to rationalize their portfolios and perform a global impact assessment in order to implement the necessary compliance changes.
- MDR's Annex I – the 'General Safety and Performance Requirements' – outlines new conditions that will need to be addressed for most CE marked MDs under the MDD. Devices which were lawfully placed on the EU market pursuant to MDD or AIMD prior to MDR date of application may continue to be made available until end of May 2025 – if the manufacturer fulfils the specific requirements drawn in the MDR; this means that MD Companies subject to transition will need to review their processes including the quality assurance, risk management and post-market product outlook. This requires a conscientious planning, implementation and review in strict compliance with the MDR.
- The MDR sets out the regulatory obligations of economic operators as it require most MD companies to update their technical documentation, clinical data and labeling ( for instance the Unique Device Identification (UDI) has to be implemented in order to to enable global device traceability throughout the entire MD manufacturer supply chain , UDI should be shown on all labels).
- The scope of medical device is broadened, covering both devices for specific medical purposes alongside devices without medical purpose such as non-medical and cosmetic devices (which previously were not regulated). For instance products for cleaning, disinfection or sterilization of devices as well as devices for the purpose of control or support or conception, hair removal lasers, liposuction equipment and contact lenses are regulated throughout the MDR.
- The MDR provides new classification rules for software (classified into classes I, IIa, IIb or III depending on the intended use of the software and a risk profile) while the software itself is considered an active medical device; also regarding software the MDR introduces the concept of interoperability to devices including software, as the ability to exchange information and use the information that has been exchanged for correct execution of specified function without changing the content of the data, and/or communicate with each other, and/or work together as intended.
- Manufacturers will need to generate and provide more in-depth clinical data to prove safety and performance claims including tighter equivalence standards. MDR provides a new understanding of clinical evaluation as being a systematic and planned process to continuously generate, collect, analyze and assess the clinical data to verify the safety and performance of a device when used as intended by the manufacturer, moreover the clinical evaluation report is required for all classes of devices and throughout the product life cycle process.
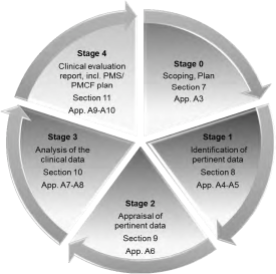
- The MDR details with greater emphasis the Quality Management System responsibilities: the Legal Manufacturer must provide a person responsible for regulatory compliance (which is a new provision explicitly defined), qualification requirements (specific education, training and experience), Provision for ability to make decision (“shall suffer no disadvantage in relation to the proper fulfilment of his duties …”) and covers responsibilities (conformity of device verified before product release, technical documentation are kept up-to-date, PMS obligations are complied with, etc)
- The MDR sets explicit provisions on manufacturers’ responsibilities for the follow-up of the quality, performance and safety of devices placed on the market. Manufacturers of all devices classes will need to report all incidents, injuries and deaths into an EU portal (Centralized Post Market Surveillance System -PMS) that will centralize data so the and user will have access to safety-related information. The timelines for serious incidents reports is shortened to 15 days from previous 30 days, while field safety corrective actions should be submitted through an electronic system.
- The MDR contains a new centralized database (EUDAMED) for Device registration, Conformity Assessment, Vigilance (incidents and recalls), Universal Device Identifier (UDI), Surveillance Reports, Clinical Investigations, Notified Bodies. MDR/IVDR UDI and device data sets provide for their registration in EUDAMED.
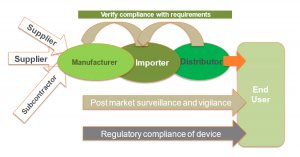
- MDR provides a strict set of general obligations to verify compliance with requirements of Economic Operators which includes the Legal Manufacturer, the EC Authorized Representative, the Importer, the Distributor and the Person who puts together [refer to Article 22 [1] or sterilizes [refer to Article 22[3] system or procedure packs.
- MDR involves also key changes as regards the NBs: as current NB designations will expire with the date of application of the MDR, all NB applicants for MDR accreditation must go through a re-designation process (observing organizational, quality management, resource and processes matters); as a result of re-designation, only few selected NBs will have a scope covering Class III high-risk devices.
- Through the MDR the NB oversight must be observed for reclassification of many medical devices to a higher risk class (for instance a new classification for reusable surgical devices is observed).
The latest MDR commitment deadlines are presented below:
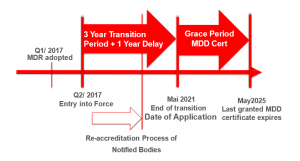




The MDs manufacturers should be advised on the onward movement towards the MDR. The MD Manufacturers of already CE certified devices under either MDD or AIMD are strongly recommended to consult their respective NBs in order to evaluate possible compliance issues, to develop a plan for a timely transition towards the MDR and also to perform an assessment of how their current MD portfolio may be impacted by the MDR. This is extremely important as not all MD design changes are anticipated or expected while this fact could likely lead to re-certification under the MDR. Let’s give a practical example: a MD manufacturer receives a new report on adverse event which will trigger a label update; as a consequence the Competent Authority (CA) may request corrective action related to the label change, this CA request will suddenly push the manufacturer forward into the MDR transition.
If you are planning your EU MDR transition or need help on MDR requirements AICROS team is here to help. Call us on: +4917621065564 or email us at: info@aicros.com
Bibliography:
- Regulation (EU) 2017/745 of the European Parliament and of the Council , April 2017
- Regulation (EU) 2020/561 of the European Parliament and of the Council of 23 April 2020 amending Regulation (EU) 2017/745 on medical devices, as regards the dates of application of certain of its provisions
- Factsheet for Manufacturers of in vitro Diagnostic Medical Devices, European Union, Nov 2018
- WHO Global Model Regulatory Framework for Medical Devices, WHO Technical Report Series, No. 1003, 2017
- Guide for Clinical Evaluation Under EU-MDR, The FDA group, Mar 2018
- The New European MDR Harmonization Effort with International Regulatory Requirements for Medical Devices, Stephan Buttron, Mar 2016